Research
DNA Replication and Genome Instability
Accurate cell division requires complete duplication of nuclear DNA, providing the daughter cell with a precise copy of the encoded genomic information. For faithful replication, the replication phases of initiation, elongation and termination must be coordinated to copy the nuclear DNA once and only once per cell cycle. Faithful replication must also retain higher order epigenetic components of chromatin that confer transcriptional regulation, three-dimensional organization, and cell identity.
DNA damage can arise as a consequence of replicative stress, a phenomenon that can be broadly defined as any impediment leading to replication fork stalling or collapse. Replication stress can be induced from endogenous sources such as DNA damage from reactive oxygen species (ROS), nucleotide pool imbalance, and repetitive DNA elements. Many chemotherapeutic agents act as exogenous sources of replication stress. By targeting DNA replication, these drugs all induce extensive DNA damage specifically in rapidly dividing tumor cells, leading to cell death.
DNA damage can arise as a consequence of replicative stress, a phenomenon that can be broadly defined as any impediment leading to replication fork stalling or collapse. Replication stress can be induced from endogenous sources such as DNA damage from reactive oxygen species (ROS), nucleotide pool imbalance, and repetitive DNA elements. Many chemotherapeutic agents act as exogenous sources of replication stress. By targeting DNA replication, these drugs all induce extensive DNA damage specifically in rapidly dividing tumor cells, leading to cell death.
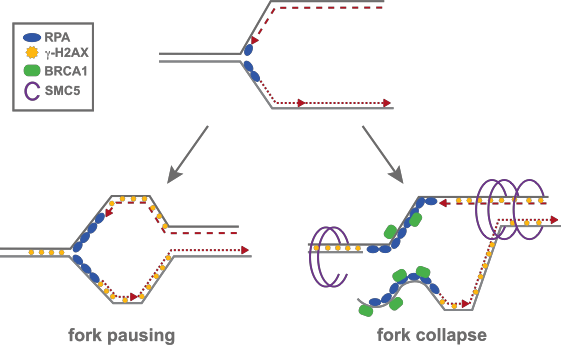
Spontaneous replication stress is also a principal source of endogenous DNA damage in cycling cells. Repair of replication damage can result in insertions, deletions and complex rearrangement events, reminiscent to those observed in cancer. Lymphocytes are particularly prone to replication damage as they undergo massive bursts of proliferation throughout development and in response to antigen stimulation, initiating the programmed DNA mutation and rearrangement processes of somatic hypermutation (SH) and class switch recombination (CSR). Replication damage may also produce secondary mutations in cancer cells, inducing mutations that promote adaptation and chemoresistance. Indeed, many proteins involved in DNA replication and cell cycle progression are dysregulated in human cancers. Thus, replication-induced DNA damage may supply both the initiating lesions responsible for tumor development but also contribute to further rearrangement and evolution of the cancer genome.
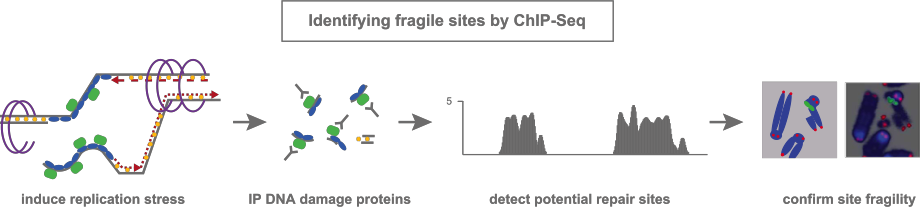
While the mechanism underlying the sensing and repair of DNA damage has been extensively studied, little is known about where damage arises during replication stress. By monitoring the association of repair proteins to DNA using chromatin precipitation followed by massive parallel sequencing (ChIP-Seq) in response to replication stress, we identified preferred genomic loci designated as early replicating fragile sites (ERFSs) that are susceptible to premature fork collapse. ERFSs are not distributed randomly throughout the genome, but associate preferentially with transcriptionally active clusters containing divergent and convergent gene pairs. Similar to CFSs, ERFSs are not DNA sequence-specific and span several hundred kilobases. Further, we show that ERFS-associated DSBs can recombine with AID dependent DSBs originating in G1 to form chromosomal translocations. Consistent with this, ERFSs are preferentially rearranged in biopsies from human B cell lymphomas. Based on these data, we propose that transcription-dependent fork collapse at ERFSs contribute to the oncogenic mechanisms of lymphomagenesis.
Current and Future studies
|
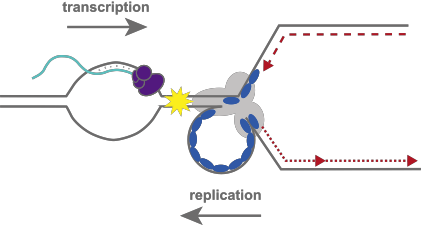
Transcription and genome instability in S phase
While transcriptional activity and replication timing have been linked to DNA breakage and genome rearrangements in a number of model systems, the tools to study these processes at endogenous loci have been elusive. ERFS provide a unique tool to understand the underlying cause of DNA fragility in response to DNA replication stress in transcriptionally active regions. Further investigation of the mechanism underlying ERFS fragility will help determine both the causes of genomic instability in the context of active transcription and replication stress, and how replication stress contributes to tumorigenesis in B lymphocytes. |
|
Mapping spontaneous and drug-induced genome rearrangements
The development of inexpensive high-throughput sequencing has provided an array of new techniques to identify genomic loci that have been mutated or undergone genome rearrangements. Previous studies mapping DNA rearrangements on a genome-wide scale have primarily occurred in immortalized—predominantly cancer-derived—cell lines or in tumor samples themselves. While informative, identifying “driver” from “passenger” mutations remains challenging. We are developing new sequencing and microscopy-based techniques to identify and characterize endogenous genomic loci harboring genome rearrangements in primary cells. Such techniques will help us to understand how mutations arise and evolve over time.
The development of inexpensive high-throughput sequencing has provided an array of new techniques to identify genomic loci that have been mutated or undergone genome rearrangements. Previous studies mapping DNA rearrangements on a genome-wide scale have primarily occurred in immortalized—predominantly cancer-derived—cell lines or in tumor samples themselves. While informative, identifying “driver” from “passenger” mutations remains challenging. We are developing new sequencing and microscopy-based techniques to identify and characterize endogenous genomic loci harboring genome rearrangements in primary cells. Such techniques will help us to understand how mutations arise and evolve over time.
